
Cancers, Vol. 12, Pages 903: Role of DNA Damage Response in Suppressing Malignant Progression of Chronic Myeloid Leukemia and Polycythemia Vera: Impact of Different Oncogenes
Cancers doi: 10.3390/cancers12040903
Authors:
Jan Stetka
Jan Gursky
Julie Liñan Velasquez
Renata Mojzikova
Pavla Vyhlidalova
Lucia Vrablova
Jiri Bartek
Vladimir Divoky
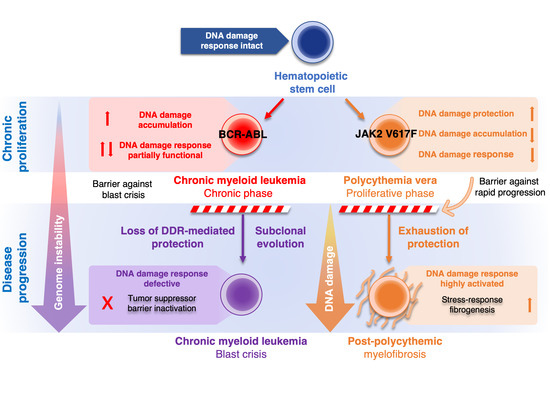
Inflammatory and oncogenic signaling, both known to challenge genome stability, are key drivers of BCR-ABL-positive chronic myeloid leukemia (CML) and JAK2 V617F-positive chronic myeloproliferative neoplasms (MPNs). Despite similarities in chronic inflammation and oncogene signaling, major differences in disease course exist. Although BCR-ABL has robust transformation potential, JAK2 V617F-positive polycythemia vera (PV) is characterized by a long and stable latent phase. These differences reflect increased genomic instability of BCR-ABL-positive CML, compared to genome-stable PV with rare cytogenetic abnormalities. Recent studies have implicated BCR-ABL in the development of a "mutator" phenotype fueled by high oxidative damage, deficiencies of DNA repair, and defective ATR-Chk1-dependent genome surveillance, providing a fertile ground for variants compromising the ATM-Chk2-p53 axis protecting chronic phase CML from blast crisis. Conversely, PV cells possess multiple JAK2 V617F-dependent protective mechanisms, which ameliorate replication stress, inflammation-mediated oxidative stress and stress-activated protein kinase signaling, all through up-regulation of RECQL5 helicase, reactive oxygen species buffering system, and DUSP1 actions. These attenuators of genome instability then protect myeloproliferative progenitors from DNA damage and create a barrier preventing cellular stress-associated myelofibrosis. Therefore, a better understanding of BCR-ABL and JAK2 V617F roles in the DNA damage response and disease pathophysiology can help to identify potential dependencies exploitable for therapeutic interventions.
Δεν υπάρχουν σχόλια:
Δημοσίευση σχολίου